
the endpoint of the titration is overshot for sale
The endpoint of titration is overshot! Does this technique error result in an increase, a decrease, or have no effect on the reported percent acetic acid in the vinegar? Explain.
Titration is a quantitative analytical volumetric technique that permits the determination of the unknown concentration of an analyte with a known concentration of titrant. This is possible because the two react in a known stoichiometric manner allowing calculation of the unknown concentration.

This website is using a security service to protect itself from online attacks. The action you just performed triggered the security solution. There are several actions that could trigger this block including submitting a certain word or phrase, a SQL command or malformed data.
Look up the pH indicator using the link in the Resources section if you are performing an acid-base titration. The pH indicator is a chemical that changes color over a given pH range. Before beginning your titration, you should have added a couple drops of the indicator, so you"ll already know which one you are using. The link under the Resources section will tell you what color change to expect when your titration is complete.
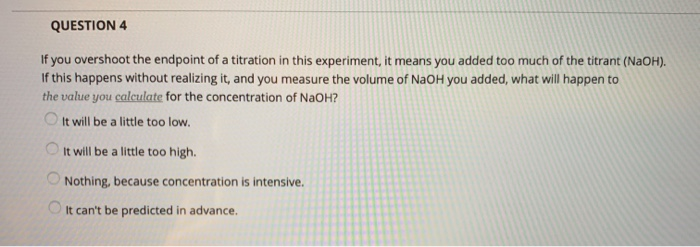
Swirl the flask of analyte while adding titrant from the buret. (The analyte is the chemical you are testing in the titration, while the titrant is the standard you are adding.) Swirling will make sure the analyte remains well-mixed so that the color change (when it occurs) is an accurate reflection of the pH of the solution.
Try placing the flask containing the analyte on a piece of paper or a Kimwipe. The white background will help you see the color change when it occurs.
Add titrant slowly. If you add titrant too quickly, you could easily overshoot the end point of your titration, at which point you may need to repeat the entire experiment.
Watch the solution closely for the color change. Once the solution begins to change color and the new color persists for at least 30 seconds, you have reached the end point of your titration.
Tips The most common mistake is to add titrant too quickly and overshoot the end point. Try doing a "quick and dirty" trial run to get an idea of how many milliliters of titrant are needed to reach the endpoint. Once you know approximately where the endpoint is, you can go back and do a more careful second trial; this time, you can add titrant quickly until you begin to approach the volume you added in the first "quick and dirty" trial and then slow down and add titrant just a drop at a time.
In Table 1, the acid–base indicators are given, which are included in this paper. Their transition intervals, and the preparation of indicator solutions are included [12, 13]. All compounds were of analytical grade. To prepare the indicator Tashiro, Methylene blue from Riedel de Haën was used. The concentrations of the indicators were chosen so that the influence of the protonation/deprotonation of the indicator is negligible, but colours were clearly detectable.
o = 0.05 mol L−1, Merck) were used. Solid borax (disodium tetraborate decahydrate), Na2B4O7·10H2O (Merck), was used to prepare a stock solution with a concentration of 0.05 mol L−1. Titrations were performed by using standard glass burettes with a reading precision of 0.025 mL (half of a scale division). A certain amount of the sample was transferred with a standard glass pipette (±0.05 mL) into a conical flask, and it was filled up with water to about 100 mL. Then the indicator was added (resulting concentrations in the sample solutions are given in Table 1), and the titration was performed. Each experiment was repeated 10 times.
Sample solutions for potentiometric titrations were prepared in the same manner in a beaker. The titrant was added either with the standard glass burette in case of high concentrations of the sample, or with a transfer pipette (10–100 ± 0.6 μL, VWR, Germany) in case of small concentrations. Titrations were followed by using a glass electrode (InLab®, Mettler Toledo, Swiss) in conjunction with a pH metre 761 Calimatic (Knick, Germany). The glass electrode was calibrated before each measurement. For each sample and concentration, 11 experiments were performed (one with each indicator).
To construct the colour maps, the titration curves were recorded by measuring the pH of the solutions with a glass electrode, and the coloured solutions where photographed in spectrophotometric cuvettes. For this, Britton-Robinson buffer solutions were prepared according to [21] with a variation of the pH in 0.4 pH increments. For each indicator, these buffer solutions were prepared and the indicator was added in the concentrations given in Table 1. The pH of each buffer solution was measured in the absence and presence of the indicator, and no detectable differences could be noted. Individual pictures were taken and the colour maps were created using the freeware PhotoScape.
3 being an amino group. Additionally, the introduction of polar substituents, such as carboxyl or sulfonic acid groups increases the solubility of the compounds. Azo dyes are indicator bases. During protonation a cation is formed, and the positive charge is delocalized, i.e., resonance between the aromatic ring and a quinoide structure (Scheme 2). The colour change is from yellow to red (or red violet).
Schematic description of the structural changes of Azo dyes during protonation/deprotonation (only one resonance structure is given for the indicator base and two for the indicator acid)
Methyl orange is an orange powder. The sodium salt is moderately soluble in water. The aqueous stock solution should contain 0.1 % of the indicator. The transition interval is from 3.0 to 4.4 (Scheme 3).
o ≈ 5 × 10−4 mol L−1). The colours of the starting solutions (v = 0 and τ = 0 in Fig. 4) differ in case of high and low concentrations, because the titrations start at different pH values. In case of high concentrations, the starting colour is of vivid red pink (pH = 2.1), changing to red orange during progressive titration. At τ ≈ 0.91 (τ: degree of titration) the solution is already orange in colour (pH = 3.1). A distinct transition takes place between τ ≈ 0.985 and τ = (0.994 ± 0.002) from orange to bright yellow (pH ≈ 3.8–4.2, titration error at first bright yellow −0.6 %). In case of small concentrations the colour of the starting solutions is already orange (pH = 3.0). The transition from orange to bright yellow takes place between τ ≈ 0.8 and τ = (0.96 ± 0.01) (pH ≈ 3.7–4.4, titration error −4 %).
o ≈ 0.001 mol L−1) with 0.1 M NaOH and Methyl orange as indicator. Similar curves are observable for H2SO4. Dots experimental points, straight lines fits of experimental results with CurTiPot
o ≈ 5 × 10−3 mol L−1, pH = 2.4), the sample solution with Methyl orange is of vivid red pink, too (Fig. 5a). But the colour changes quickly to red orange during progressive titration. Already at τ ≈ 0.5 (pH = 3.8) the solution is yellow orange in colour. The transition takes place between τ ≈ 0.62 and τ = (0.90 ± 0.03) from yellow orange to bright yellow (pH ≈ 4.7, titration error −10 %). In case of low concentrations (C
o ≈ 5 × 10−4 mol L−1, pH = 3.3) the colour of the starting solution is already yellow orange (Fig. 5b). The transition from yellow orange to bright yellow is smeared out, and bright yellow is reached at τ = 0.83 ± 0.04 (pH ≥ 4.5, titration error −17 %).
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Methyl orange as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
o ≈ 5 × 10−4 mol L−1) are brilliant yellow before the addition of hydrochloric acid (pH = 9.2 and pH = 9.0 for high and low concentrations, respectively). In case of high concentrations, the transition from yellow to yellow orange (first orange undertone) is observable at τ = 1.012 ± 0.004 (pH = 4.3, titration error +1.2 %), but the colour changes gradually to orange, red orange and at least vivid red pink when continuing the addition of hydrochloric acid (i.e., large excess, pH < 3.0, cf. Figure 6a). In case of low concentrations, the transition from yellow to yellow orange is hardly visible at τ = 1.17 ± 0.06 (pH = 4.3, titration error +17 %), a real transition to orange and red orange is only obtainable, if an excess of hydrochloric acid is added (cf. Fig. 6b).
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Methyl orange as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Methyl red is a very common indicator for titrations of strong acids with strong bases. It is a dark red violet crystalline powder. In contrast to Methyl orange, it is sparingly soluble in water, and a stock solution should contain 0.02 % of the indicator in a 60 % ethanol water mixture. The transition interval is from pH 4.4 to 6.2 (Scheme 4). In the literature [13], the colour transition is given from yellow (base) to red (acid), but as it is shown in Fig. 7, a better description for the colour of the acid is fuchsia.
o ≈ 0.001 mol L−1) with 0.1 M NaOH and Methyl red as indicator. Similar curves are observable for H2SO4. Dots experimental points, straight lines fits of experimental results with CurTiPot
In case of high and low concentrations of strong acids, the sample solutions with Methyl red are of brilliant fuchsia colour (pH = 2.1 and pH = 3.0 for C
o ≈ 0.001 mol L−1, respectively). This colour does not change markedly during ongoing titration (Fig. 7a, b). The transition from fuchsia to yellow is very sharp. Only very close to the transition point (τ > 0.997 and τ > 0.98 in case of high and low concentrations, respectively, pH ≈ 4.5), the fuchsia colour starts getting lighter. At τ ≈ 1.000 (τ = 0.999 ± 0.002, high concentration, and τ = 1.00 ± 0.02, low concentration, pH ≈ 6–7) the complete solution is yellow. In both cases the systematic error is negligible, as we will discuss later in detail. However, it must be mentioned here, that the yellow colour vanishes with time, and the colour of the solutions returns to fuchsia because carbon dioxide from ambient air dissolves, and this decreases the pH of the solution. The rate of the regeneration of the fuchsia colour depends strongly on the experimental and ambient conditions, e.g. the return of the colour is very fast when the solution is heavily shaken. Hence, it is not serious to give a certain time span in which the colour should stay, although, this is often done in textbooks. In praxis, this leads very often to the fact that undergraduate students read the end point too late.
In case of a weaker acid like oxalic acid, the transition is not as sharp as for strong acids (cf. Fig. 8a, b). We can see here, that both solutions are fuchsia before titration starts. The brilliant fuchsia turns pale at τ > 0.90 (pH > 4.7). At τ ≈ 0.98 (pH = 5.5) the pale fuchsia turns to red orange, and the transition to clear yellow is obtained at τ = 0.990 ± 0.003 and τ = 0.99 ± 0.01 (pH = 5.7) for high and low concentrations of oxalic acid, respectively (titration error −1 %).
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Methyl red as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
In case of borax a sharp transition is observable; here the colour changes from yellow to brilliant fuchsia (Fig. 9a, b). At τ ≈ 0.99 (high concentration) and τ ≈ 0.98 (low concentration) (pH ≈ 6.0) the solutions start getting slightly orange (very weak orange undertone), and at τ = 1.000 ± 0.003 and τ = 1.00 ± 0.01 the transition to fuchsia for low and high concentrations is obtained, respectively (pH = 5.2). In both cases, the titration error is negligible.
To give sharper endpoint colour changes, Methyl red can be mixed with a dye insensitive to pH changes like Methylene blue (Scheme 5). This mixture is called Tashiro. The transition interval is that of Methyl red, the colour changes from violet (mixture of blue and brilliant fuchsia) to green (mixture of yellow and blue). At the transition point (pH = pK
Congo red, as the disodium salt, is a red powder. It is soluble in water, and the aqueous stock solution should contain 0.1 % of the indicator. The transition interval is from 3.0 to 5.0. This compound has a comparatively high salt error. It is a bisazo compound, resulting in a colour change different to that described above (cf. Scheme 6). In the literature [12], the colour transition is given from blue (acid) to red (base), but as shown in Fig. 11, it is better to describe the colour transition as blue violet to orange.
Sample solutions of high and low concentrations of hydrochloric acid and sulfuric acid with Congo red are pale blue violet (pH = 2.1 and pH = 3.0 for high and low concentrations, respectively, cf. Fig. 11a, b). In case of high concentrations the colour starts getting brighter already at τ ≈ 0.93 (pH ≈ 3.2). Between τ ≈ 0.98 and τ ≈ 0.99 (pH ≈ 3.7–4.0) the transition takes place from violet to red violet, and the solutions become orange above τ = 0.993 ± 0.002 (pH = 4.2, titration error −0.7 %). In case of low concentrations the transition from violet to red violet and at least to orange is observable between τ ≈ 0.8 and τ = (0.94 ± 0.02) (pH ≈ 3.7–4.3, titration error −6 %).
In case of oxalic acid, both titrations start with a pale blue violet colour (pH = 2.4 and pH = 3.3 for high and low concentration, respectively) (Fig. 12a, b). In case of high concentrations, at τ ≈ 0.7 (pH ≈ 3.8–4.0) it starts getting brighter and the solution becomes more violet. Between τ ≈ 0.80 and τ ≈ 0.85 the transition from red violet to orange red is observable, a clear orange is obtained at τ = 0.87 ± 0.02 (pH = 4.6, titration error −13 %). In case of low concentration, the transition interval is smeared out over a larger range of the titration degree; the solution starts getting brighter already about τ ≈ 0.5 (pH ≈ 3.8), and a clear orange is obtained already at τ = 0.77 ± 0.04 (pH = 4.5, titration error −23 %).
Figure 13a and b show the titration curves for high and low concentrations of borax with HCl and Congo red as indicator. In both cases, the solutions are orange before adding the HCl. In case of high concentration, the transition from orange via red violet to violet is rather sharp, it starts at τ ≈ 1.01 (pH = 4.5), and the transition to blue violet is obtained at τ = 1.014 ± 0.004 (pH = 4.0, titration error +1.4 %). In case of small concentration, the transition starts later at τ ≈ 1.05 (pH = 4.5), and the transition from red violet to blue violet is not as sharp as in case of high concentrations. A clear blue violet is obtained at τ = 1.15 ± 0.02 (pH = 4.0), i.e., the titration error is +15 %.
The Phthaleins (Scheme 7) are synthesised by heating phthalic anhydride with a derivative of phenol. All Phthaleins are rather insoluble in water but soluble in ethanol. In the acidic form most of the Phthaleins are colourless (lactone form), while in alkaline media a coloured quinoide structure is formed because of the opening of the lactone ring, i.e., most Phthaleins are monochromic indicators (Scheme 8).
Structural changes of Phthaleins during protonation/deprotonation (only one resonance structure is shown for the indicator acid and one resonance structure for the indicator base)
Structural changes of Phthaleins in strong alkaline solutions during protonation/deprotonation (pH > 13, only one resonance structure is shown for each species)
Phenolphthalein, one of the most popular indicators, can be considered as the basic compound of Phthaleins (Scheme 10). It is a white crystalline powder; stock solutions contain 0.05–0.1 % of the indicator in an ethanol water mixture of 50–70 % ethanol. The transition interval of phenolphthalein is from 8.0 to 10.
Solutions of high and low concentrations of strong acids as well as of weak acids appear colourless, when Phenolphthalein is added (see Figs. 14a, b, 15a, b). Under the conditions we consider here (concentration of Phenolphthalein is given in the experimental section, transition point at pH ≈ 8.5), for strong acids a sudden colour change to faint pink is obtained at τ = 1.003 ± 0.002 and τ = 1.02 ± 0.02 (high and low concentration, respectively). It is of outmost importance to avoid all colour influences from outside, meaning that the background is covered with white paper. The faint colour fades with time until the solution appears colourless again due to the influence of ambient air (cf. Methyl red). If the solution appears dark pink, the endpoint is overshot. Phenolphthalein is a suitable indicator for the titration of weak acids with a strong base as it can be seen in case of oxalic acid (Fig. 15a, b). Here, the colour turns to faint pink at τ = 1.000 ± 0.003 (high concentration) and τ = 1.01 ± 0.01 (low concentration).
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Phenolphthalein as indicator. Similar curves are observable for HCl. Dots experimental points, straight lines fits of experimental results with CurTiPot
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Phenolphthalein as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
In contrast, Phenolphthalein is absolutely inappropriate as an indicator for the titration of weak bases with a strong acid, as it is depicted in Fig. 16a, b for borax. Although the sample solutions are pink, this colour fades quickly during ongoing titration, and the systematic deviation exceeds −40 % (τ = 0.54 ± 0.12 for high concentration, τ = 0.53 ± 0.08 for low concentrations).
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Phenolphthalein as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Thymolphthalein is a white crystalline powder; the structure is rather similar to Phenolphthalein with the exception that the two phenol groups are replaced by two thymol groups (Scheme 11). A stock solution contains 0.1 % of the indicator in an 80 % ethanol solution. Like phenolphthalein, it is a single-colour indicator. The transition range lies between pH 9.7 and 10.5.
Thymolphthalein turns from colourless to blue, but it has to be mentioned here, that it is only a fade blue, the transition is rather difficult to see and the influence of carbon dioxide from ambient air is rather pronounced due to the high pH values. In that range, the addition of about 5 × 10−5 mol L−1 of H3O+ ions leads to a change in pH of about 0.3 units. In case of strong acids (Fig. 17a, b), the first very faint blue colour is obtained between pH ≈ 9.7 and 10.0, i.e., τ = 1.009 ± 0.003 (high concentration, titration error +0.9 %), and τ = 1.06 ± 0.02 (low concentration, titration error +6 %). As in case of Phenolphthalein, it is important to avoid all colour influences from outside, and the progress of titration should be controlled with a white background.
o ≈ 0.001 mol L−1) with 0.1 M NaOH and Thymolphthalein as indicator. Similar curves are observable for H2SO4. Dots experimental points, straight lines fits of experimental results with CurTiPot
Similar results are obtainable with a weak acid like oxalic acid, as it is depicted in Fig. 18a, b. The first very faint blue colour is obtained between pH ≈ 9.7 and 10.0, i.e., τ = 1.006 ± 0.002 (titration error +0.6 %) in case of high concentration, and τ = 1.06 ± 0.02 (titration error +6 %) in case of low concentration.
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Thymolphthalein as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Weak bases like borax are not titratable using Thymolphthalein as indicator. Here, already the sample solutions are colourless, as it is shown in Fig. 19a, b.
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Thymolphthalein as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Sulfonephthaleins (Scheme 12) are closely related to the Phthaleins and are formed by the reaction of o-sulfobenzoic acid anhydride with a phenol. They are sparingly soluble in water in the biprotonated form, whereas the monosodium salt is soluble in water. The structure contains the less stable sulfone ring which is opened already in acidic solutions. This leads to a quinoide structure, and the sulphonphthaleins are coloured in the acidic form, too. Sulfonephthaleins can be deprotonated in two steps [22]:
Bromophenol blue is an orange or light brown powder. Aqueous stock solution should contain 0.1 % of the monosodium salt of the indicator. The transition interval is from 3.0 to 4.6 (Scheme 14).
The solutions of high and low concentrations of hydrochloric acid and sulfuric acid with Bromophenol blue are pale yellow in colour (pH = 2.1 and pH = 3.0 in case of high and low concentrations, respectively, cf. Fig. 20a, b). In case of high concentrations, around τ = 0.97–0.98 (pH ≈ 3.5–3.7) the solutions start getting greenish yellow to grey and then to pale blue. The most striking colour change takes place at τ = 0.989 ± 0.003 (pH ≈ 4.0) the colour changes to blue purple resulting in a titration error of −1.1 %. During ongoing addition of the titrant, a smeared out colour change from blue purple to purple was observed. At lower concentrations, a faint greenish yellow colour is already visible at τ = 0.7 (pH ≈ 3.5), but the transition to blue purple takes place at τ = 0.94 ± 0.01 (pH ≈ 4.2), resulting in a titration error of −6 %.
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Bromophenol blue as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
In case of oxalic acid, the sample solution with high concentration of the acid (pH = 2.4) shows a pale yellow colour, too (Fig. 21a). Already at τ ≈ 0.6 (pH ≈ 3.6), the solution gets greenish yellow, and at τ ≈ 0.7 (pH ≈ 3.8–4.0) the colour changes via grey to pale blue. A clear transition to blue purple takes place at τ = 0.87 ± 0.02 (pH = 4.6), i.e., a titration error of −13 %. In case of low concentration of oxalic acid (Fig. 21b), the starting solution is greenish yellow in colour (pH = 3.3), and the colour changes slightly right from the beginning of the titration. Between τ ≈ 0.4 and τ ≈ 0.8, the colour turns gradually from pale blue to blue. The transition to blue purple takes place at τ = 0.86 ± 0.05 giving a titration error of −14 %.
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Bromophenol blue as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Solutions of high and low concentrations of borax with Bromophenol blue as indicator are purple at the beginning, changing gradually to blue purple during ongoing titration (Fig. 22a, b). The blue colour starts degrading at τ ≈ 0.99 (high concentration) and τ ≈ 1.00 (small concentration) (pH ≈ 5.0–4.5). A transition from pale blue via grey to greenish yellow is observable between τ ≈ 1.00 and τ ≈ 1.03 for high concentrations, and between τ ≈ 1.01 and τ ≈ 1.15 for low concentrations, respectively (pH ≈ 4.5–3.2). If grey colour (pH ≈ 4.0) is taken for the determination of the transition point, the systematic errors are +1 and +8 % for high and low concentrations, respectively. In both cases, one will not end up with a clear pale yellow colour.
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Bromophenol blue as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Bromocresol green in the acidic form is a green powder. The monosodium salt is a dark red amorphous hydrate which becomes colourless at about 190 °C. The aqueous stock solution should contain 0.04 % of the indicator. The transition interval lies between pH 3.8 and 5.6 (Scheme 15).
Sample solutions with high concentrations of strong acids with Bromocresol green as indicator are yellow in colour (pH = 2.1, Fig. 23a). The colour changes at τ > 0.91 (pH > 3.1) to become greenish yellow, but a clear colour change from green to bright blue green is observed at τ = 0.999 ± 0.002 (pH ≈ 5.0, titration error −0.1 %), i.e., the systematic error is negligible. Also in case of small concentrations of strong acids, a sharp colour change is observed (Fig. 23b). The solutions are slightly greenish yellow right on from the beginning of the titration (pH = 3.0). Bright blue green solutions are observed for τ = 1.00 ± 0.01.
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Bromocresol green as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
A quite different result is obtainable in case of high and low concentrations of a weak acid like oxalic acid. Starting with a yellow solution, in case of high concentration (pH = 2.4) it becomes greenish yellow, soon; in case of low concentration we already start with a greenish yellow solution (pH = 3.3). The colour change is smeared out from turquoise to bright blue green between τ ≈ 0.70 and τ = (0.97 ± 0.01) (pH = 4.0–5.4) for high concentrations, and between τ ≈ 0.75 and τ = (0.91 ± 0.03) (pH = 4.4–5.0) for low concentrations (i.e., the titration errors are −3 and −9 %, cf. Fig. 24a, b).
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Bromocresol green as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Solutions of borax in high and low concentration are bright blue green (pH = 9.2 and pH = 9.0 for high and low concentration, respectively), when Bromocresol green is added. Here, with ongoing addition of hydrochloric acid, the transition from blue green via turquoise to greenish yellow is rather sharp, and at τ = 1.009 ± 0.002 (high concentrations) and τ = 1.05 ± 0.02 (low concentrations) the greenish yellow colour is established (pH = 4.2, titration errors +0.9 and +5 %). In case of high concentrations, the colour will change until a yellow solution results, but this needs a markedly excess of hydrochloric acid (τ ≈ 1.1, pH ≈ 3.5). In case of small concentrations the solutions are still greenish yellow at τ ≈ 1.2 (pH ≈ 3.6) (cf. Fig. 25).
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Bromocresol green as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Thymol blue forms dark green crystals. A stock solution can be prepared by dissolving 100 mg of the indicator in a solution containing 0.001 mol L−1 sodium hydroxide. This indicator shows two transition regions: one at pH 1.2–2.8 from red orange to greenish yellow, and one at pH 8.0–9.6 from greenish yellow to blue (Scheme 16).
Only in case of high concentrations of strong acids, a smeared transition from red orange to greenish yellow can be observed at the beginning of the titration (cf. Fig. 26a), because the sample solutions have a pH = 2.1. In case of low concentrations of strong acids one already starts with a greenish yellow solution (pH = 3.0, cf. Fig. 26b). The same holds true for high and low concentrations of oxalic acid (pH = 2.4 and pH = 3.3, cf. Fig. 27a, b). In all cases, a sharp transition from greenish yellow to blue is obtained at the following degrees of titration: τ = 1.000 ± 0.003 (strong acid, high concentration), τ = 1.01 ± 0.02 (strong acid, low concentration), τ = 1.002 ± 0.003 (H2C2O4, high concentration), τ = 1.01 ± 0.01 (H2C2O4, low concentration), i.e., the titration error is negligible. But also here we can observe the influence of carbon dioxide (as it was already discussed in case of Methyl red). The blue colour turns quickly to green.
o ≈ 0.001 mol L−1) with 0.1 M NaOH and Thymol blue as indicator. Similar curves are observable for H2SO4. Dots experimental points, straight lines fits of experimental results with CurTiPot
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Thymol blue as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
In case of borax the sample solutions are blue, but the colour turns quickly to greenish yellow (τ ≈ 0.4), and a definite determination of a transition point is impossible (cf. Fig. 28a, b).
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Thymol blue as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Cresol red forms dark green–red crystalline powder. The stock solution should contain 0.04 % of the indicator. As in case of thymol blue, in very strong acidic solutions (pH < 1.8), the quinoide structure is protonated and the zwitter ion is red in colour. At pH values above 1.8, the deprotonated quinoide system is yellow, and the transition from yellow to vivid violet is between 7.2 and 8.8 (Scheme 17).
In all cases considered here, only the transition from yellow to vivid violet (or vice versa in case of borax) is considered. The transition is very sharp, very close to the transition point the solutions appears pale pink to nearly colourless (pH ≈ 7.3–7.8), (cf. Figs. 29a, b, 30a, b, 31a, b). As it was discussed already for other indicators, the influence of carbon dioxide from ambient air is very strong, during the titration of acids with a strong base. The colour turns quickly back to yellow. For all the three acids considered here in high and low concentration the titration error is negligible. The end points are obtained at the following degrees of titration: τ = 1.003 ± 0.003 (strong acid, high concentration), τ = 1.00 ± 0.01 (strong acid, low concentration); τ = 0.999 ± 0.002 (H2C2O4, high concentration), τ = 1.00 ± 0.02 (H2C2O4, low concentration).
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Cresol red as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
In case of borax we start with the vivid violet solutions (Fig. 31a, b). At τ ≈ 0.8 (pH ≈ 8.2) the colour turns to pale pink (nearly colourless). A clear transition from colourless to yellow is obtained at τ = 0.95 ± 0.01 (titration error −5 %) in case of high concentration and τ = 0.92 ± 0.02 (titration error −8 %) in case of low concentration (pH = 7.5).
Neutral red is one representative of other aromatic compounds suitable as acid base indicators. It is a dark green, nearly black powder moderately soluble in water and ethanol. Stock solutions contain 0.01 % of the indicator in an ethanol water mixture of 50–70 % ethanol. The transition interval ranges from 6.8 to 8.0. At pH values above 7.4, the uncharged molecule is the dominant species [24]. At lower pH values the cation is dominant (Scheme 18). The salt error of the compound is very small.
Structures of indicator acid and indicator base of Neutral red (only one resonance structure is displayed for the indicator base, two possible resonance structures are given for the indicator acid)
In case of strong and weak acids in both, high and low concentrations, the sample solutions are fuchsia, and a sharp transition from fuchsia via salmon to mustard yellow is observable (Figs. 32a, b, 33a, b), but solutions turn rapidly to fuchsia again under ambient conditions (see discussions above). For all the three acids considered here in high and low concentration the titration error is negligible. The end points are obtained at the following degrees of titration: τ = 1.001 ± 0.003 (strong acid, high concentration), τ = 1.00 ± 0.02 (strong, acid, low concentration); τ = 0.999 ± 0.002 (H2C2O4, high concentration), τ = 1.01 ± 0.02 (H2C2O4, low concentration).
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and neutral red as indicator. Similar curves are observable for HCl. Dots experimental points, straight lines fits of experimental results with CurTiPot
o ≈ 5 × 10−4 mol L−1) with 0.1 M NaOH and Neutral red as indicator. Dots: experimental points, straight lines: fits of experimental results with CurTiPot
In case of borax titrations, it is much more difficult to determine the end point, because the transition starts already at τ ≈ 0.95 (high concentration) and τ ≈ 0.90 (low concentration) (pH ≈ 7.5), and the colour changes from salmon via pale pink to fuchsia. A clear fuchsia is obtained at τ = 0.993 ± 0.004 (high concentration) and τ = 0.99 ± 0.02 (low concentration) (pH = 6.8, Fig. 34a, b).
o ≈ 5 × 10−4 mol L−1) with 0.1 M HCl and Neutral red as indicator. Dots experimental points, straight lines fits of experimental results with CurTiPot
Manual titrations are time consuming and can be inaccurate at times due to human error. Not to mention, important data can get easily lost due to improper tracking methods. The Thermo Scientific Orion Star T900 Series Automated Titrators are designed to make performing titrations easier, more reliable, and more reproducible than manual titrations.
These auto titrators expand the number of ions and compounds that can be measured beyond direct electrode analysis and offer dynamic process controls that adjust the titration to optimize analysis results.
Manual titration can be a time consuming and frustrating process. Watch how easy it is to find the endpoint, reproduce your workflow, and optimize your results. The auto-filling burette helps to minimize the handling of corrosive materials. Use of an auto titrator well help ensure a safer, more efficient lab.
A water treatment plant in the midwestern United States that ran up to 10,000 titrations each year improved their workflow. Learn how streamlining the workflow using an Orion Star Automated Titrator for low-level alkalinity titrations benefited the lab.
In this white paper, you’ll learn about the dispense accuracy and precision of the Orion Star T900 Series Automated Titrators. We’ll demonstrate that our auto titrators exceed well-established industry precision and accuracy specifications, providing users with greater confidence in their titration applications.
Streamline your manual titration workflows and increase efficiency and repeatability with an Orion Star T900 Series Automated Titrator. Review the applications chart below to find out if an auto titrator is for you.
We are currently unable to offer solutions for Karl Fischer, amperometric, stat, and dead stop titrations. For other questions please contact customer support to be connected to your local sales representative.
Get our top 10 tips for performing automated titrations, and methodologies for common uses of an auto titrator. Discover how to perform an acid/base titration for orange juice, water and petroleum in this ebook.
Performing manual titrations can be extremely tedious, requiring the operator to stand in one place, watching minuscule droplets drip into a sample container and diligently waiting for the color change or other endpoint indictor to occur before starting the process all over again, for possibly hours and hours of repeated sample titrations.
An automatic titrator allows you to start the titration and then walk away from the titrator to perform other tasks or tests while the titrator takes care of the titrant addition, endpoint detection and results calculations automatically without any involvement from the operator.
Manual titrations typically use a non-certified, Class B or Class A burette with stopcock to add doses of titrant to the sample. The operator uses the stopcock to start and stop the additions of titrant into the sample, often one drop at a time, until the endpoint is reached.
The precision of these additions, especially near the endpoint, is primarily determined by the operator’s skill level, experience and focus on the task at hand. All too commonly, an operator can allow too much titrant to flow out of the burette into the sample and overshoot the endpoint, requiring them to perform the entire titration all over again. Even highly skilled and experienced operators can be limited in the precision of their manual titration results by the last few drops of titrant, since each drop can vary in volume.
When using an automatic titrator, the titration is performed using a high-accuracy titrant delivery system that controls all titrant additions into the sample and will adjust the dose rate as it detects the endpoint approaching.
Once the endpoint is reached, the titrator uses the precisely measured volume of titrant added to the sample to automatically calculate the concentration results for the sample. The operator’s skill level, experience and focus on the task at hand is no longer a factor in the overall accuracy of the titration results and the possibility of missing the endpoint is greatly reduced.
Typically calculating titrations in the lab is done by calculating the sample concentration after the endpoint has been reached. There are many points in this calculation where human error can get in the way of consistent, reproducible results.
When using an auto titrator, it will automatically calculate the sample concentration from the entered parameters. Plus, on the automatic titrator, electrode, titrant and titration setup parameters can be saved as a method, so the exact same settings are used for each titration. These methods can be transferred between titrators for consistent procedures to be used on multiple titrators or multiple labs. This way you can save time with repeat titrations by running the exact same parameters each time without having to reenter any information.
Instead of manually logging the titration results in a notebook or scrap of paper, an auto titrator will automatically save the titration results in the data log with time and date stamp.

This website is using a security service to protect itself from online attacks. The action you just performed triggered the security solution. There are several actions that could trigger this block including submitting a certain word or phrase, a SQL command or malformed data.

A smooth titration graph and receiving results back from your titrator does not always guarantee that they are accurate. Always review your results, your calculations, and quality checks.
It is important when reviewing titration results to ask yourself, “Does this result make sense?”. This is especially the case when switching methodologies, i.e. from manual titrations to automatic titrations. It can be alarming to see an unfamiliar number, but it is important to take a closer look at your specifications to determine the cause of deviation. The four most important parts of a specification to examine are the units, significant digits, the range, and the methodology used to originate the specifications. We will talk about the significance of each.
There are many different ways to represent the results of titration. Units of specifications can vary, but specifications are commonly represented in %, ppm (mg/L or mg/g), or simply mL (milliliters) of titrant used to determine the endpoint. Results can be customized in the method options to match those of the specifications in more cases. Results that vary from the original specification by a common factor, like 10 or a 1000 are most commonly attributed to a difference in units and can be easily adjusted.
The other part of the unit is the analyte, or what specific chemical is being tested. In order to ensure that the results match the specification, it needs to be clear what form of the analyte is being represented.
In the example of a salt titration, some customers prefer that results be represented in sodium chloride, whereas some specifications represent the result in just chloride. The titration is exactly the same for both units, but the calculated results differ because of the difference in molar mass between sodium chloride and chloride. In the case of acidity, titration results are normally represented in units of the predominant acid. A specification that is written in % lactic acid, would differ greatly from a result represented in % citric acid. This is true, not only because of the differences in molar mass, but also the reaction ratio between the titrant and the specific acid. These deviations can easily be corrected in the method so that the results match the specification.
Usually, specifications are written as a range. For example, as discussed above, the sample should contain between 2.0% to 2.5% sodium chloride. Having a wide degree of tolerance in the range of a specification allows some leeway for time savings and sources of error. A very narrow specification range will require good lab technique and adherence to best practices to ensure that the results achieved are accurate.Specifications should be written with these limitations in mind, as well as being based upon the expected concentration of the analyte.The following are the AOAC guidelines for percent recovery and percent relative standard deviation (%RSD) based on the expected analyte concentration.
When possible, it is always a good idea to understand the methodology used to originate the specification. Different methods have varying degrees of accuracy and unique potential interferences. When moving from one methodology to another, i.e. from manual titration to automatic titration, there may be a slight difference in results. In the case of manual titrations, the endpoint is often overshot and the results can vary between operators. This leads to slightly higher numbers than the actual results. Specifications may need to be reevaluated when switching methodologies to improve the accuracy of the specification.
In summary, familiarity with specifications will make interpreting the results of titration much easier. Implementing quality checks, discussed below, will help to ensure that titration results are accurate.
Quality checks are typically used by labs to ensure the accuracy of their reported results. By implementing quality checks into your analysis procedure, you will not only have peace of mind but, you will also have documentation in case the results are called into question. There are several types of quality checks, but we will focus on blanks, laboratory control spikes, duplicates, and matrix spikes.
A blank is a sample of your solvent that is carried through the sample preparation procedure and then titrated to ensure there is no interference or contamination from the analysis procedure. Remember that any titrant that is being dosed is being calculated into the results. Titrant that reacts with your solvent should not be included in sample results and can be factored out in the method options of the titrator.
A Laboratory Control Spike, abbreviated LCS, is a standard of known analyte concentration that is carried through the sample analysis procedure to evaluate the accuracy of the titration method. For example, if we were titrating the concentration of nickel in a plating bath, we would use a nickel standard of verified concentration to validate the accuracy of the testing procedure. The AOAC has published guidelines on percent recovery as it pertains to the concentration of analyte which is listed above. Laboratory Control Spikes can be used to validate new methods or to ensure the continued accuracy of current methods.
A duplicate is a sample that is carried through sample analysis as two independent samples to ensure that the results of the method are repeatable. Often when validating new methods, multiple replicates are analyzed to determine the relative standard deviation (RSD) between samples. The AOAC also has guidelines for %RSD as they pertain to the analyte concentration.
A sample matrix spike, often abbreviated as MS, is a sample that is duplicated and to which a standard of known concentration is added to one of the replicates. Matrix spikes are useful in determining if the other components of your sample are interfering with the methods.
Note: Quality Checks are not limited to the titrator. Pipettes and balances can also be checked using reference material to ensure that they are maintaining their precision and accuracy. Quality checks can be performed as frequently as preferred, but are typically done at least daily or at the beginning of every shift.

A titration is a volumetric technique in which a solution of one reactant (the titrant) is added to a solution of a second reactant (the "analyte") until the equivalence point is reached. The equivalence point is the point at which titrant has been added in exactly the right quantity to react stoichiometrically with the analyten (when moles of titrant = moles of analyte). If either the titrant or analyte is colored, the equivalence point is evident from the disappearance of color as the reactants are consumed. Otherwise, an indicator may be added which has an "endpoint" (changes color) at the equivalence point, or the equivalence point may be determined from a titration curve. The amount of added titrant is determined from its concentration and volume:
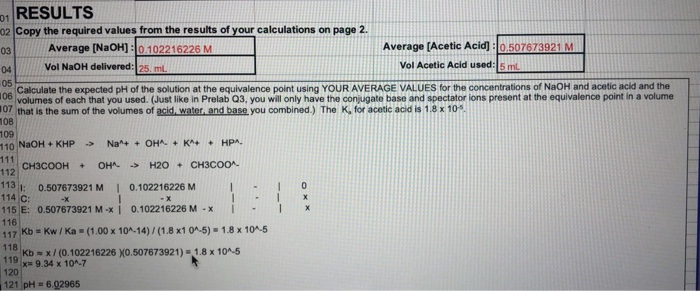
A measured volume of the solution to be titrated, in this case, colorless aqueous acetic acid, CH3COOH(aq) is placed in a beaker. The colorless sodium hydroxide NaOH(aq), which is the titrant, is added carefully by means of a buret. The volume of titrant added can then be determined by reading the level of liquid in the buret before and after titration. This reading can usually be estimated to the nearest hundredth of a milliliter, so precise additions of titrant can be made rapidly.
Figure \(\PageIndex{1}\):The titration setup initially, before titrant (NaOH) has been added. NaOH is held in the burett, which is positioned above the beaker of acetic acid. Titrant (NaOH) is added until it neutralizes all of the analyte (acetic acid). This is called the equivalence point. Note: Unlike the picture, both substances are actually clear but are blue for visibility purposes in the picture.
As the first few milliliters of titrant flow into the flask, some indicator briefly changes to pink, but returns to colorless rapidly. This is due to a large excess of acetic acid. The limiting reagent NaOH is entirely consumed.
The added indicator changes to pink when the titration is complete, indicating that all of the aqueous acetic acid has been consumed by NaOH(aq). The reaction which occurs is
Reaction of acetic acid and sodium hydroxide to give acetate ion, sodium ion and water. The reaction is shown in terms of stick and ball diagram of each species.
Eventually, all the acetic acid is consumed. Addition of even a fraction of a drop of titrant produces a lasting pink color due to unreacted NaOH in the flask. The color change that occurs at the endpoint of the indicator signals that all the acetic acid has been consumed, so we have reached the equivalence point of the titration. If slightly more NaOH solution were added, there would be an excess and the color of the solution in the flask would get much darker. The endpoint appears suddenly, and care must be taken not to overshoot the endpoint.
After the titration has reached the endpoint, a final volume is read from the buret. Using the initial and final reading, the volume added can be determined quite precisely:
Figure \(\PageIndex{2}\)The figure above shows a completed titration, where the equivalence point has been reached. NaOH (the titrant) has neutralized all of the Acetic Acid, leaving Acetate in the beaker. At this point, the moles of NaOH added is equivalent to the moles of acetic acid initially in the beaker.
The object of a titration is always to add just the amount of titrant needed to consume exactly the amount of substance being titrated. In the NaOH—CH3COOH reaction Eq. \(\ref{2}\), the equivalence point occurs when an equal molar amount of NaOH has been added from the graduated cylinder for every mole of CH3COOH originally in the titration flask. That is, at the equivalence point the ratio of the amount of NaOH, added to the amount of CH3COOH consumed must equal the stoichiometric ratio
Titration is often used to determine the concentration of a solution. In many cases it is not a simple matter to obtain a pure substance, weigh it accurately, and dissolve it in a volumetric flask as was done in Example 1 of Solution Concentrations. NaOH, for example, combines rapidly with H2O and CO2 from the air, and so even a freshly prepared sample of solid NaOH will not be pure. Its weight would change continuously as CO2(g) and H2O(g) were absorbed. Hydrogen chloride (HCl) is a gas at ordinary temperatures and pressures, making it very difficult to handle or weigh. Aqueous solutions of both of these substances must be standardized; that is, their concentrations must be determined by titration.
A sample of pure potassium hydrogen phthalate (KHC8H4O4) weighing 0.3421 g is dissolved in distilled water. Titration of the sample requires 27.03 ml NaOH(aq). The titration reaction is
To calculate concentration, we need to know the amount of NaOH and the volume of solution in which it is dissolved. The former quantity could be obtained via a stoichiometric ratio from the amount of KHC8H4O4, and that amount can be obtained from the mass
By far the most common use of titrations is in determining unknowns, that is, in determining the concentration or amount of substance in a sample about which we initially knew nothing. The next example involves an unknown that many persons encounter every day.
Vitamin C tablets contain ascorbic acid (C6H8O6) and a starch “filler” which holds them together. To determine how much vitamin C is present, a tablet can be dissolved in water andwith sodium hydroxide solution, NaOH(aq). The equation is
If titration of a dissolved vitamin C tablet requires 16.85 cm³ of 0.1038 M NaOH, how accurate is the claim on the label of the bottle that each tablet contains 300 mg of vitamin C?
The 308.0 mg obtained in this example is in reasonably close agreement with the manufacturer’s claim of 300 mg. The tablets are stamped out by machines, not weighed individually, and so some variation is expected.

I would like to say hello to all of you. Let"s examine the question. So here any, which is standardized by using KH p the first K HP is we used standardized work and you do so so in this case, what has done that? The polarity of K K HP is so point hola. The volume of K HP is 25 men and the period reading is 25 minutes. The three trials were done and the trading was good. The volume is 25 mil only. The volume off spirit trading is what it is. There is also 25 minutes called for. We have to calculate the polarity of any of them. We know that the modularity of KH p is equal to the modularity of any which is greater than the volume. The K H p V no is in the same direction. 25 minutes to here is the volume. This is what it is. Too cold. There are 25 items on this page. I"m not in contact with anyone. If we calculate the modularity of any, it will be equal to 0. This is the word that is calculated, which is modularity. So the second experiment orders did that. The hydration was sexy because another solution of any was taken to hear more clarity of any. So does it call 2.1250? Our volume of spirit was traded off the bureau. Let"s just be bitter about trading. The sample of its cell is about 2010 normal and the spirit reading is a call to 44 liters. For that 10 liter 50 liter of water, you have to do that. He said the total volume will be 16. So the volume of the seal is the same as the stent Plus 15. Now, the question is how many moles are required for the tight ration. Now we have to calculate the number of moles, since the volume is 44, and this much more clarity is required. A cold is the number of moles. Both the values are given more clarity and volume. By volume, it was multiplied by 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 and 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 888-349-8884 That leader is also a twin. Similarity is. 1250. By volume is 44 and by entries is 3. This is equal to five more if you calculate it. So this is the number of moles of water and in which, and again, so the modularity of excel is asked to find out the polarity of it still can be found again by using the same formula similarity of any which is larger than the volume Of anything. It is the same as modularity of excel, Multiplied by volume. How much does it call? Here it is a culture for the poor polarity of its sale, which is 60 mil. Is it cold if you calculate the polarity of it? .092 Molars. It"s okay. It was asked to find out the number of moles in the cell that was present in this. So the third question is, the number of moles that exhale. The formula can be used to find out if this can be found again. Yes, right. The polarity is calculated by summing up the volume of the game. But industry is -3. This is called 2.005 five if you calculate it. The number of moles is in malta. Thank you for taking the time to respond.

It is a cost-effective testing method because it does not involve any specialist kit beyond the burette and other glassware containers, plus a colour indicator and the titrant.
The automated system dispenses more precise, smaller doses of titrant, and many devices can also provide dynamic dosing where the amounts get progressively smaller when approaching an endpoint.
It is a time-saving approach that reduces the likelihood of human error during the titration process along with helping to prevent any overshooting of the endpoint.
Therefore if the testing reveals poor results for salt content, then anything produced during that time will either become waste or have to be put in quarantine.
Real-time measurement of seasoning while making snack food gives production line staff the opportunity to intervene and make adjustments without losing whole runs of a product.

You are viewing Bowen or Logan Overshots for sale by Pickett Oilfield, LLC. We have an extensive range of good used or rebuilt Overshots along with the accessories. Call us for your fishing tool needs. For more information contact us by phone at sales@pickettoilfield.com.
PickettOilfield.com offers prospective buyers an extensive selection of quality new, used, and refurbished Oilfield Equipment at competitive prices, including Overshots.
Overshots are a tool used on the downhole process during the fishing operations. It engages the tube or tool on the outside surface. A Grapple or other piece of equipment that is similar attaches onto the overshot and grabs the fish.

This article was co-authored by Bess Ruff, MA. Bess Ruff is a Geography PhD student at Florida State University. She received her MA in Environmental Science and Management from the University of California, Santa Barbara in 2016. She has conducted survey work for marine spatial planning projects in the Caribbean and provided research support as a graduate fellow for the Sustainable Fisheries Group.
wikiHow marks an article as reader-approved once it receives enough positive feedback. In this case, 86% of readers who voted found the article helpful, earning it our reader-approved status.



 8613371530291
8613371530291