
jin rongsheng uc irvine made in china

Structure and function of bacterial toxins and receptors; synaptic proteins; protein complexes; protein-protein and protein-ligand interactions; X-ray crystallography; high-throughput screening
Our research group is dedicated to understand the molecular basis of human diseases using structural biology, which allows us to visualize how proteins function or malfunction at the atomic level. Our current research is focused on three areas: (1) exploring the molecular mechanisms underlying the toxic and therapeutic functions of botulinum neurotoxins (BoNTs), which will help to develop effective anti-BoNT strategies and improve clinical applications of BoNT; (2) characterizing the structures of Clostridium difficile (C. diff) toxins (TcdA and TcdB) and their interactions with host receptors, and understanding how they contribute to the disease of Clostridium difficile infection (CDI) that tops the CDC’s list of urgent threats; (3) advancing mechanistic understanding of ion channels, receptors, and signaling molecules in the nervous system, which will facilitate the design and improvement of therapeutic agents for the treatment of some psychological and neurological disorders. We are also developing cutting-edge small molecule high-throughput screening (HTS) assays based on our understanding of the structure and function of these disease-related proteins, which may lead to novel chemical probes and/or drug candidates for basic research and therapeutic application.
Lam, K. H., Guo, Z., Krez, N., Matsui, T., Perry, K., Weisemann, J., Rummel, A., Bowen, M. E. & Jin, R. A viral-fusion-peptide-like molecular switch drives membrane insertion of botulinum neurotoxin A1. Nat Commun 9, 5367 (2018) doi: 10.1038/s41467-018-07789-4.
Chen, P., Tao, L., Liu, Z., Dong, M. & Jin, R. Structural insight into Wnt signaling inhibition by Clostridium difficile toxin B. FEBS J (2018) doi: 10.1111/febs.14681.
Chen, P., Tao, L., Wang, T., Zhang, J., He, A., Lam, K. H., Liu, Z., He, X., Perry, K., Dong, M*. & Jin, R*. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360, 664-669 (2018) (*corresponding authors) doi: 10.1126/science.aar1999. PMCID: PMC6231499
Lam, K. H., Sikorra, S., Weisemann, J., Maatsch, H., Perry, K., Rummel, A., Binz, T. & Jin, R. Structural and biochemical characterization of the protease domain of the mosaic botulinum neurotoxin type HA. Pathog Dis 76 (2018) doi: 10.1093/femspd/fty044. PMCID: PMC5961070
Silva, D. A., Stewart, L., Lam, K. H., Jin, R. & Baker, D. Structures and disulfide cross-linking of de novo designed therapeutic mini-proteins. FEBS J 285, 1783-1785 (2018) doi: 10.1111/febs.14394. PMCID: PMC6001749
Lam, K. H., Qi, R., Liu, S., Kroh, A., Yao, G., Perry, K., Rummel, A. & Jin, R. The hypothetical protein P47 of Clostridium botulinum E1 strain Beluga has a structural topology similar to bactericidal/permeability-increasing protein. Toxicon 147, 19-26 (2018) doi: 10.1016/j.toxicon.2017.10.012. PMCID: PMC5902665
Chevalier, A., Silva, D.A., Rocklin, G.J., Hicks, D.R., Vergara, R., Murapa, P., Bernard, S.M., Zhang, L., Lam, K.H., Yao, G., Bahl, C.D., Miyashita, S.I., Goreshnik, I., Fuller, J.T., Koday, M.T., Jenkins, C.M., Colvin, T., Carter, L., Bohn, A., Bryan, C.M., Fernández-Velasco, D.A., Stewart, L., Dong, M., Huang, X., Jin, R., Wilson, I.A., Fuller, D.H. & Baker, D. Massively parallel de novo protein design for targeted therapeutics. Nature 550(7674):74-79 (2017) doi: 10.1038/nature23912. PMCID: PMC5802399
Yao, G., Lam, K.H., Weisemann, J., Peng, L., Krez, N., Perry, K., Shoemaker, C.B., Dong, M., Rummel, A. & Jin, R. A camelid single-domain antibody neutralizes botulinum neurotoxin A by blocking host receptor binding. Sci Rep. 7;7(1):7438. (2017) doi: 10.1038/s41598-017-07457-5. PMCID: PMC5547058
Yao, G., Lam, K.H., Perry, K., Weisemann, J., Rummel, A. & Jin, R. Crystal Structure of the Receptor-Binding Domain of Botulinum Neurotoxin Type HA, Also Known as Type FA or H. Toxins (Basel) 9, 93 (2017) doi: 10.3390/toxins9030093. PMCID: PMC5371848
Yao, G., Zhang, S., Mahrhold, S., Lam, K. H., Stern, D., Bagramyan, K., Perry, K., Kalkum, M., Rummel, A.*, Dong, M.* & Jin, R.* N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat Struct Mol Biol 23 (7):656-662 (2016) (*corresponding authors) doi: 10.1038/nsmb.3245. PMCID: PMC5033645
Lee, K., Lam, K. H., Kruel, A. M., Mahrhold, S., Perry, K., Cheng, L. W., Rummel, A. & Jin, R. Inhibiting oral intoxication of botulinum neurotoxin A complex by carbohydrate receptor mimics. Toxicon 107, 43-49 (2015) doi: 10.1016/j.toxicon.2015.08.003. PMCID: PMC4658216
Lam, K.H. & Jin, R. Architecture of the botulinum neurotoxin complex: a molecular machine for protection and delivery. Current Opinion in Structural Biology 31:89-95 (2015) doi: 10.1016/j.sbi.2015.03.013. PMCID: PMC4476938
Lam, K.H., Yao, G. & Jin, R. Diverse binding modes, same goal: The receptor recognition mechanism of botulinum neurotoxin. Progress in Biophysics and Molecular Biology 117(2-3):225-31 (2015) doi: 10.1016/j.pbiomolbio.2015.02.004. PMCID: PMC4417461
Lam, T.I., Stanker, L.H., Lee, K., Jin, R. & Cheng, L.W. Translocation of botulinum neurotoxin serotype A and associated proteins across the intestinal epithelia. Cellular Microbiology 17(8):1133-1143 (2015) doi: 10.1111/cmi.12424. PMCID: PMC4610714
Matsui, T.*, Gu, S., Lam, K.H., Carter, L.G., Rummel, A., Mathews, II. & Jin, R.* Structural Basis of the pH-Dependent Assembly of a Botulinum Neurotoxin Complex. J. Mol. Biol. 426(22):3773-3782 (2014) doi: 10.1016/j.jmb.2014.09.009. (*corresponding authors) PMCID: PMC4252799
Lee, K., Zhong, X., Gu, S., Kruel, A.M., Dorner, M.B., Perry, K., Rummel, A., Dong, M. & Jin, R. Molecular basis for disruption of E-cadherin adhesion by botulinum neurotoxin A complex. Science 344(6190):1405-1410 (2014) doi: 10.1126/science.1253823. PMCID: PMC4164303
Lee, K., Lam, K.H., Kruel, A.M., Perry, K., Rummel, A. and Jin, R. High-resolution crystal structure of HA33 of botulinum neurotoxin type B progenitor toxin complex. Biochem. Biophys. Res. Commun. 446(2):568-573 (2014) doi: 10.1016/j.bbrc.2014.03.008. PMCID: PMC4020412
Yao, Y., Lee, K., Gu, S., Lam, K.H. & Jin, R. Botulinum Neurotoxin A Complex Recognizes Host Carbohydrates through Its Hemagglutinin Component, Toxins (Basel) 6(2):624-635 (2014) doi: 10.3390/toxins6020624. PMCID: PMC3942755
Lee, K., Gu, S., Jin, L., Le, T.T.N., Cheng, L.W., Strotmeier, J., Kruel, A.M., Yao, G., Perry, K., Rummel, A.* & Jin, R.* Structure of a Bimodular Botulinum Neurotoxin Complex Provides Insights into Its Oral Toxicity. PLoS Pathog. 9(10): e1003690 (2013) doi:10.1371/journal.ppat.1003690. (*corresponding authors) PMCID: PMC3795040
Zong, Y. and Jin, R. Structural mechanisms of the agrin-LRP4-MuSK signaling pathway in neuromuscular junction differentiation. Cell. Mol. Life Sci. 70(17):3077-88 (2013) doi: 10.1007/s00018-012-1209-9. PMCID: PMC4627850
Gu, S. and Jin, R. Assembly and function of the botulinum neurotoxin progenitor complex. Curr. Top. Microbiol. Immunol. 364:21-44 (2013) doi: 10.1007/978-3-642-33570-9_2. PMCID: PMC3875173
Gu, S., Rumpel, S., Zhou, J., Strotmeier, J., Bigalke, H., Perry, K., Shoemaker, C.B., Rummel, A. & Jin, R. Botulinum neurotoxin is shielded by NTNHA in an interlocked complex. Science 335(6071):977-81 (2012) doi: 10.1126/science.1214270. PMCID: PMC3545708
Zong, Y., Zhang, B., Gu, S., Lee, K., Zhou, J., Yao, G., Figueiredo, D., Perry, K., Mei, L.* & Jin, R.* Structural basis of neuron-specific regulation of postsynaptic differentiation. Gene & Development 26:247-258 (2012) doi: 10.1101/gad.180885.111. (*corresponding authors) PMCID: PMC3278892
Yao, G., Zong, Y., Gu, S., Zhou, J., Xu, H., Mathews, II. & Jin, R. Crystal structure of the glutamate receptor GluA1 amino-terminal domain. Biochem. J. 438(2):255-63 (2011) doi: 10.1042/BJ20110801. PMCID: PMC3296483
Strotmeier, J., Gu, S., Jutzi, S., Mahrhold, S., Zhou, J., Pich, A., Eichner, T., Bigalke, H., Rummel, A.*, Jin, R.* & Binz, T*. The biological activity of botulinum neurotoxin type C is dependent upon novel types of ganglioside binding sites. Mol. Microbiol. 81(1):143-56 (2011) doi: 10.1111/j.1365-2958.2011.07682.x. Epub 2011 Jun 2. (*corresponding authors)
Strotmeier, J., Lee, K., Völker, A.K., Mahrhold, S., Zong, Y., Zeiser, J., Zhou, J., Pich, A., Bigalke, H., Binz, T., Rummel, A.* & Jin, R.* Botulinum neurotoxin serotype D attacks neurons via two carbohydrate-binding sites in a ganglioside-dependent manner. Biochem. J. 431(2):207-16 (2010) (*corresponding authors)
Jin, R.*, Singh, S.K., Gu, S., Furukawa, H., Sobolevsky, A.I., Zhou, J., Jin, Y. & Gouaux E.* Crystal structure and association behavior of the GluR2 amino-terminal domain. EMBO J. 28(12):1812-23 (2009) (*corresponding authors) PMCID: PMC2699365
Kumar, J., Schuck. P., Jin, R. & Mayer, M.L. The N-terminal domain of GluR6-subtype glutamate receptor ion channels. Nat. Struct. Mol. Biol. 16(6):631-8 (2009) PMCID: PMC2729365
Jin, R., Rummel, A., Binz, T. & Brunger, A.T. Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature 444:1092-5 (2006)
Jin, R., Clark, S., Weeks, A.M., Dudman, J.T., Gouaux, E. & Partin, K.M. Mechanism of positive allosteric modulators acting on AMPA receptors. J. Neurosci. 25(39):9027-36 (2005)
Jin, R., Junutula, J.R., Matern, H.T., Ervin, K.E., Scheller, R.H. & Brunger, A.T. Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J. 24:2064-74 (2005)
Jin, R., Bank, T., Mayer, M. L., Traynelis, S. & Gouaux, E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 6(8):803-10 (2003)

Structure of the glucosyltransferase domain of TcdA in complex with RhoA provides insights into substrate recognition. Sci Rep. 2022 05 30; 12(1):9028.
Jahid S, Ortega JA, Vuong LM, Acquistapace IM, Hachey SJ, Flesher JL, La Serra MA, Brindani N, La Sala G, Manigrasso J, Arencibia JM, Bertozzi SM, Summa M, Bertorelli R, Armirotti A, Jin R, Liu Z, Chen CF, Edwards R, Hughes CCW, De Vivo M, Ganesan AK. PMID: 35385746; PMCID: PMC9127750.
Probing the structure and function of the protease domain of botulinum neurotoxins using single-domain antibodies. PLoS Pathog. 2022 01; 18(1):e1010169.
Chen P, Zeng J, Liu Z, Thaker H, Wang S, Tian S, Zhang J, Tao L, Gutierrez CB, Xing L, Gerhard R, Huang L, Dong M, Jin R. PMID: 34145250; PMCID: PMC8213806.
Structural Insights into Rational Design of Single-Domain Antibody-Based Antitoxins against Botulinum Neurotoxins. Cell Rep. 2020 02 25; 30(8):2526-2539.e6.
Chen P, Lam KH, Liu Z, Mindlin FA, Chen B, Gutierrez CB, Huang L, Zhang Y, Hamza T, Feng H, Matsui T, Bowen ME, Perry K, Jin R. PMID: 31308519; PMCID: PMC6684407.
The hypothetical protein P47 of Clostridium botulinum E1 strain Beluga has a structural topology similar to bactericidal/permeability-increasing protein. Toxicon. 2018 Jun 01; 147:19-26.
High-resolution crystal structure of HA33 of botulinum neurotoxin type B progenitor toxin complex. Biochem Biophys Res Commun. 2014 Apr 04; 446(2):568-73.

Botulism is caused when the botulinum neurotoxin (BoNT) inhibits the release of a neurotransmitter. The disease can be caused by eating toxin-contaminated food, but how the BoNT protein survives the digestive tract and reaches the bloodstream has been a mystery. Last year, a group led by Rongsheng Jin of the University of California, Irvine, demonstrated how a protein called nontoxic nonhemagglutinin (NTNHA) binds to and shields BoNT to protect it from digestive proteases. Jin and colleagues have now used electron microscopy and X-ray crystallography to study a complex of BoNT, NTNHA, and three hemagglutinin proteins that play a role in getting BoNT past intestinal cells to the blood (PLoS Pathog. 2013, DOI: 10.1371/journal.ppat.1003690). The researchers find that the 760-kilodalton complex evokes the construction of the Apollo lunar lander, with BoNT and NTNHA on top and the hemagglutinins forming “legs,” which are the parts that interact with intestinal epithelial cells. The legs land on and bind to sugars on the cells, facilitating passage of BoNT. Jin and colleagues find that dosing mice with a monosaccharide can reduce BoNT toxicity, suggesting a way to prevent—but not treat—botulism. The full protein complex could also point to ways to deliver protein drugs orally.

Part of the answer lies in the molecules that carry the toxin through the body. These carriers, which are produced along with the toxin by the Clostridium botulinum bacterium, protect the toxin as it travels through the hostile environment of the gastroinstetinal tract, and help it bust through the intestinal wall and into the bloodstream.
In a paper published in the latest issue of the journal Science, researchers report they have figured out the crystal structure of one of the toxin"s molecular carriers. This structure helps scientists understand how the toxin is able to sneak through the lining of the intestine and into the blood.
"The whole complex looks just like the Apollo lunar module," says Rongsheng Jin, biophysicist at the University of California, Irvine and lead author on the paper. He compares the complex to the toxin"s "landing gear".
This complex helps the toxin in two ways. First, as Jin implies, it lands on and then binds to the cells lining the inside of the intestine. "If they cannot stick there, [the toxin] would just go through and end up in the toilet," says Jin.
By the time a patient presents with botulism poisoning, the toxin is already in their blood. John Mark Carter, research chemist in the produce safety and microbiology research unit at the Department of Agriculture, says that at this point "intestinal therapy doesn"t really help."
However, Jin says that understanding how these vehicles work could help scientists devise better ways to deliver drugs to the body. "We can make these proteins in the lab, assemble them into a vehicle and use them to deliver drugs to the intestine," he says.

CSPG4 is a large highly glycosylated single transmembrane protein (~251 kDa). Its extracellular domain was predicted to contain a signal peptide, two laminin G motifs, and 15 consecutive CSPG repeats1a). Our initial efforts using the recombinant full extracellular domain of human CSPG4 (residues 30–2204, referred to as CSPG4ECD) and TcdB1 holotoxin (VPI10463 strain) were hampered by the structural flexibility of TcdB and CSPG4ECD employing cross-linking mass spectrometry (XL-MS) using the MS-cleavable cross-linker dihydrazide sulfoxide (DHSO)1a, b and Source Data). When forming a complex, acidic residues in TcdB1 and CSPG4ECD that have Cα-Cα distances within 35 Å can be cross-linked by DHSO, and the resulting cross-linked peptides could be identified using multistage mass spectrometry (MSn)
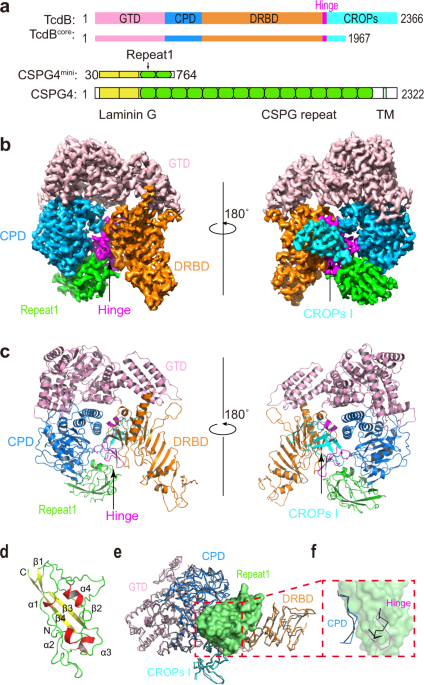
a Schematic diagrams showing the domain structures of TcdB and CSPG4, as well as the domain boundaries for TcdBcore and CSPG4mini used for cryo-EM studies. GTD: glucosyltransferase domain, CPD: cysteine protease domain, DRBD: delivery and receptor-binding domain, CROPs: combined repetitive oligopeptides domain, Hinge: a key fragment between the DRBD and CROPs that mediates structural communications among all four domains of TcdB. CSPG4 is composed of two predicted laminin G domains, 15 CSPG repeats, a transmembrane domain (TM), and a cytosolic region. b The 3.17 Å resolution cryo-EM map of the TcdBcore–Repeat1 complex segmented and colored as shown in a. c Cartoon representation of the structure of the TcdBcore–Repeat1 complex that is shown in similar orientations and color schemes as that in b. d The structure of Repeat1 of CSPG4 with the disulfide bond shown as sticks. e The structure of the TcdBcore–Repeat1 complex was superimposed to TcdB holotoxin (PDB: 6OQ5). The Repeat1-bound TcdB is colored as shown in a and the unliganded TcdB is colored black with its CROPs II–IV omitted for clarity. The TcdB-bound Repeat1 is shown as a green surface model. f Repeat1 triggers local structural changes in the CPD and hinge of TcdB upon binding. For clarity, only residues 569–577 in the CPD and residues 1803–1812 in the hinge are shown in the context of Repeat1 (green surface).
We identified a total of 263 unique DHSO cross-linked peptides of the TcdB1–CSPG4ECD complex (Supplementary Table 1), representing 18 inter-protein and 245 intra-protein (167 in TcdB1 and 78 in CSPG4ECD) cross-links. The intra-molecular cross-links in TcdB1 show good correlations with the crystal structure of TcdB1 holotoxin that we recently reported1c). The rest four pairs of cross-links suggested that the laminin G domains of CSPG4 may adopt flexible conformations and could transiently move within ~35 Å of the CPD or DRBD of TcdB, because the same residues (e.g., E92/E93) in this region of CSPG4 could be cross-linked to amino acids on the CPD and DRBD of TcdB that are 97 Å away from each other (Supplementary Table 1). Guided by the XL-MS results, we analyzed interactions between a number of fragments of TcdB1 and CSPG4 and their biochemical behaviors, and narrowed down a fragment of TcdB1 (residues 1–1967, referred to as TcdBcore) that contains the GTD, CPD, DRBD, and the first unit of CROPs (termed CROPs I), which could robustly bind to an N-terminal CSPG4 fragment composed of two laminin G motifs and first two CSPG repeats (residues 30–764, referred to as CSPG4mini) (Fig. 1a and Supplementary Table 2).
We successfully obtained a stable complex composed of TcdBcore and CSPG4mini, which was used for cryo-EM study (Supplementary Fig. 2a–d). The preliminary data analysis yielded a 3.4 Å resolution structure for the TcdBcore–CSPG4mini complex, which revealed that CSPG4mini binds to a groove in TcdB that is surrounded by the CPD, DRBD, hinge, and CROPs I (Supplementary Fig. 2e, f), which is consistent with our XL-MS studies. 3D variability analysis indicated that the distal region in the DRBD of TcdB and the N-terminal two laminin G motifs of CSPG4mini were highly flexible, which hindered us from obtaining a high-resolution map for de novo model building. Notably, these flexible regions in TcdB and CSPG4 were outside the complex interface. Therefore, we could improve the resolution by using a smaller box size during particle picking to focus on the TcdB–CSPG4 interface. With a focused refinement, we were able to further improve the density map to 3.17 Å resolution that allowed de novo model building for CSPG4, while the TcdB structure was built using the crystal structure of TcdB holotoxin as a model1b, c and Supplementary Fig. 2g, h). Structure determination statistics and representative density maps for the protein complex were shown in Supplementary Table 3 and Supplementary Fig. 3.
The structure of the TcdB–CSPG4 complex reveals that the first CSPG repeat of CSPG4 (termed Repeat1, residues 410–551) is mainly responsible for TcdB binding, while the rest of CSPG4 pointing away from the toxin (Supplementary Fig. 2f). Repeat1 has a compact structure consisting of a four-strand β sheet and 4 short α helices, which are connected by intermittent loops and stabilized by a disulfide bridge (Fig. 1d). Despite its small size, Repeat1 directly interacts with many amino acids that are dispersed across over 1300 residues on the primary sequence of TcdB, including the CPD, DRBD, hinge, and CROPs (Fig. 1b, c). All these TcdB residues converge spatially to form a composite binding site for Repeat1 involving an extensive interaction network and burying a large molecular interface between them (∼2715.5 Å2) (Fig. 2a–c). This unusually complex binding mode, especially the involvement of the CPD, is unexpected, because it was previously believed that the receptor binding of TcdB is carried out by the DRBD and the CROPs
More detailed structural analysis showed that the TcdB-binding surface in Repeat1 could be divided into three subsites (Fig. 2c). The site-1 of Repeat1 (residues 448–457) binds to the CPD via hydrogen bonds, charge–charge interaction, as well as a large patch of hydrophobic interactions (Fig. 2d and Supplementary Table 4). The site-2 of Repeat1 (residues 466–503) binds to the hinge of TcdB involving mainly hydrophobic interaction and two hydrogen bonds, and also interacts with the CROPs I with a hydrogen bond (Fig. 2e and Supplementary Table 4). The site-3 of Repeat1 is composed of two separated areas including residues 457–466 and an additional residue (R527) in a nearby loop. It binds to a composite interface in TcdB, which is composed of residues in the CPD, DRBD, and hinge (Fig. 2f and Supplementary Table 4). CSPG4 is predicted to have 15 N-linked glycosylation site with one in Repeat1 (N427) and a single chondroitin sulfate modification at S995
The overall structure of the CSPG4-bound TcdBcore is similar to the crystal structure of TcdB holotoxin with a root mean square deviation between comparable Cα atoms about 1.06 Å (Fig. 1e)1f). It is worth noting that the hinge is located at a strategic site in TcdB communicating with all four major domains, and the CROPs of TcdB adopts dynamic conformations relative to the rest of the toxin
We further carried out real-time analysis of the kinetics of TcdB–CSPG4 interactions using bio-layer interferometry (BLI). For this study, we first designed a recombinant CSPG4 Repeat1 that is fused to the N-terminus of the Fc fragment of a human immunoglobulin G1 (Repeat1-Fc). Based on the structural modeling, the Fc fragment in Repeat1-Fc does not interfere with TcdB binding, and provide a convenient way for immobilization of Repeat1-Fc to the biosensors. We found that TcdB1 recognized Repeat1-Fc with a high affinity (dissociation constant, Kd ~15.2 nM) (Supplementary Fig. 4a). Notably, Repeat1-Fc binds to TcdB with a relatively slow on-rate (kon ~7.06 × 103 M−1 s−1), which is likely due to organization of multiple structural units in TcdB to form the composite binding site for CSPG4. Nevertheless, once Repeat1 is engaged with TcdB, the complex is very stable as evidence by their slow binding off-rate (koff ~1.08 × 10−4 s−1).
Since TcdB1 and TcdB2 have different primary sequences and pathogenicity, we carried out structure-based sequence analysis between them focusing on the CSPG4-binding site. Remarkably, the key amino acids consisting the composite CSPG4-binding site are nearly identical between TcdB1 and TcdB2, even though these residues scatter across multiple TcdB domains (Fig. 2g). It is worth noting that the hinge region has large sequence variations among TcdB isoforms, and the hypervariable sequences in this region are believed to contribute to differences in toxicity and antigenicity of TcdB2 and other variantsTcdB1 with L1809TcdB2 and V1816TcdB1 with I1816TcdB2. The only other difference is N1850TcdB1 in the CROPs I that forms a hydrogen bond with K503 of CSPG4 is replaced with K1850TcdB2. Nevertheless, our BLI binding studies showed that TcdB2 binds to Repeat1-Fc with a high affinity that is even slightly better than TcdB1 (Kd ~5.4 nM, kon ~8.34 × 103 M−1 s−1, koff ~4.63 × 10−5 s−1) (Supplementary Fig. 4b). Therefore, the three residue substitutions in the CSPG4-binding site are well tolerated in TcdB2. These data demonstrate that the CSPG4-binding mode is conserved between TcdB1 and TcdB2.
We next carried out structure-guided mutagenesis of TcdB1 and CSPG4 to validate the binding interface and to define loss-of-function mutations in TcdB that could selectively abolish CSPG4 binding. We designed and characterized nine mutations of TcdB1 holotoxin, where the key CSPG4-binding residues in the CPD (L563G/I566G, S567E, Y621A, or Y603G), the hinge (D1812G, V1816G/L1818G, or F1823G/I1825G/M1831G), the DRBD (N1758A), or the CROPs I (N1850A) were mutated (Supplementary Fig. 5). These TcdB1 mutants showed reduced binding to HeLa cells expressing endogenous CSPG43a). TcdB-N1758A and N1850A showed the least reduction of binding, suggesting that these two mutations, located in the DRBD and the CROPs respectively, have relatively weaker impact on TcdB–CSPG4 interactions compared with mutations in the CPD or the hinge. We then designed three combinational mutations of TcdB to simultaneously disrupt the anchoring points for CSPG4 in both the CPD and the hinge, including S567E/D1812G, Y603G/D1812G, and S567E/Y603G/D1812G, and found them largely abolished binding of TcdB to cells. Similar results were confirmed using pull-down assays with Repeat1-Fc as the bait and TcdB variants as preys (Supplementary Fig. 6a). We also designed and characterized variants of CSPG4 Repeat1 that carried site-specific mutations in the TcdB-binding interface, including mutations in site-1 (R450G, E448A, W449G, W449D, Q453A, E448A/W449D, R450G/Q453A), site-2 (L497G, L497D, L497G/D498G), and site-3 (D457G, R464A/S466G) (Supplementary Fig. 7). These mutations effectively disrupted the binding of TcdB holotoxin to Repeat1 based on pull-down assays (Supplementary Fig. 6b).
a The indicated TcdB mutants were tested for binding to cells. Purified WT and mutated TcdB (10 nM) were incubated with WT or CSPG4−/− HeLa cells. Cells were washed three times by PBS, harvested, and cell lysates were analyzed by immunoblot detecting TcdB. Actin served as a loading control. The sensitivity of CSPG4−/− (b) and WT (c) HeLa cells to mutated TcdB was examined using the standard cytopathic cell-rounding assay. Error bars indicate mean ± sd (n = 3 biologically independent experiments). d The ratios of CR50 values on CSPG4−/− vs. WT HeLa cells from b and c were calculated and plotted, reflecting the fold-of-change in reduction of toxicity on CSPG4−/− cells compared with WT cells. n = 3 for all groups. The upper and lower bounds of boxes indicate the maximum and minimum values of each group. The middle lines indicate the median values of each group. p values by t-test: *p ≤ 0.05.
We further examined how these TcdB mutations effect CSPG4-mediated cytopathic toxicity at functional levels using standard cell-rounding assays, where TcdB entry would inactivate Rho GTPases and cause the characteristic cell-rounding phenotype50), which is utilized to compare the potency of TcdB variants on the wild-type (WT) HeLa cells that express both CSPG4 and FZDs or the CSPG4 knockout (KO) HeLa cells. As shown in Fig. 3b, all 12 mutant TcdB1 induced cell-rounding with potencies similar to TcdB1 on CSPG4 KO cells, demonstrating that these mutations were properly folded and did not affect FZD-mediated binding and entry of toxins. In contrast, these mutant toxins showed various reduced potencies on WT HeLa cells compared with TcdB1 (Fig. 3c). More specifically, WT TcdB1 showed over 600-fold reduced toxicity on CSPG4 KO cells compared with WT cells, while the toxicity of TcdB1 variants carrying L563G/I566G, D1812G, V1816G/L1818G, F1823G/I1825G/M1831G, and the three combinational mutations were similar on CSPG4 KO cells and WT cells (CR50 ratio ~1.1–1.3), demonstrating that these mutations effectively and selectively eliminated CSPG4-mediated toxicity on cells (Fig. 3d).
Given our extensive structural, in vitro, and ex vivo data demonstrating the role of CSPG4 as a TcdB receptor, we sought to determine the contribution of CSPG4 to TcdB1 and TcdB2 pathogenicity and its relationship with FZD in vivo using two complementary approaches that were custom designed for TcdB2 and TcdB1, respectively.
We next carried out histological analysis of cecum and colon tissues. There was bloody fluid accumulation in tissues dissected from WT mice after infection, whereas there was much less fluid accumulation in tissues from CSPG4 KO mice (Fig. 4a). We further carried out histological analysis with paraffin-embedded cecum tissue sections (Fig. 4b), which were scored based on disruption of the epithelium, hemorrhagic congestion, submucosal edema, and inflammatory cell infiltration, on a scale of 0–3 (normal, mild, moderate, or severe, Fig. 4c). Infection induced extensive disruption of the epithelium and inflammatory cell infiltration, as well as severe hemorrhagic congestion and mucosal edema on WT mice (Fig. 4c). CSPG4 KO mice showed only moderate levels of epithelial damage and inflammatory cell infiltration, and mild to no hemorrhagic congestion and submucosal edema (Fig. 4b, c). Furthermore, TcdB2 induced extensive loss of tight junction in the cecum epithelium from WT mice based on immunofluorescence staining for a tight junction marker Claudin-3, while it was largely intact in CSPG4 KO mice (Fig. 4d). We observed similar results when we carried out infection experiments using a ten-fold lower dose of C. difficile spores (1 × 104), which did not result in death of mice and thus allowed us to harvest cecum tissues 90 h after infection (Supplementary Fig. 8c–e). Analysis of feces indicated similar levels of C. difficile colonization and toxin titer in WT and CSPG4 KO mice (Supplementary Fig. 8c). Taken together, these results demonstrated that CSPG4 is a major receptor for the epidemic TcdB2 in vivo. The residual toxicity of TcdB2 in CSPG4 KO mice indicates that TcdB2 may have unknown low affinity receptor(s) that remains to be further evaluated.
a Three groups of infection experiment were performed: mock to WT (n = 4); M7404, tcdA− to WT mice (n = 8); and M7404, tcdA− to CSPG4−/− mice (n = 9). The representative cecum and colon of infected mice that were harvested at 48 h. The harvested cecum was processed with hematoxylin and eosin staining (scale bar represents 100 µm, mock n = 4, C. difficile to WT n = 4, C. difficile to CSPG4−/− n = 5) (b), scored based on inflammatory cell infiltration, hemorrhagic congestion, epithelial disruption, and submucosal edema (c), and subjected to immunofluorescence staining by epithelial cell junction marker Claudin-3 (scale bar represents 50 µm, mock n = 3, C. difficile to WT n = 3, C. difficile to CSPG4−/− n = 3) (d). In c, error bars indicate mean ± SEM (mock n = 4, C. difficile to WT n = 4, C. difficile to CSPG4−/− n = 5). p values were calculated by post hoc analysis of a one-way ANOVA using Holm-Sidak’s test for multiple comparisons: ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05. The exact p values are presented in the accompanying source data.
TcdB1 can be simultaneously bound by CSPG4 and FZD as demonstrated by our cryo-EM structure of the TcdB–CSPG4 complex and the crystal structure of a TcdB–FZD complex, which was confirmed by a pull-down experiment (Supplementary Fig. 8f)5a). To investigate the relationship of these two receptors for TcdB1, we resorted to three structure-based rationally designed TcdB1 mutants as molecular tools, which carry site-specific mutations to selectively knockout its binding capacity to CSPG4, FZD, or both. Based on the mutagenesis studies described above, we chose to use TcdBS567E/Y603G/D1812G as a representative CSPG4 binding deficient TcdB mutant (TcdBCSPG4−). We previously already developed a FZD-binding deficient TcdB variant that carries mutations in the FZD-binding site (TcdBGFE)FZD−/CSPG4−).
a A structural model of TcdB holotoxin with CSPG4 and FZD bound at two independent sites. The model is built based on superposition of the structures of TcdB1 holotoxin (PDB: 6OQ5), the TcdB–FZD complex (PDB: 6C0B), and the TcdB–CSPG4 complex (this work). b–d The indicated TcdB mutants or the control PBS was injected into the cecum of CD1 mice in vivo. The cecum tissues were harvested 6 h later and subjected to histological analysis with representative images (scale bars represent 100 µm, PBS n = 4, TcdB n = 5, TcdBGFE n = 5, and TcdBFZD−/CSPG4− n = 5, TcdBCSPG4− n = 5) (b), immunostaining analysis for the tight junction marker Claudin-3 (scale bars represent 50 µm, PBS n = 3, TcdB n = 3, TcdBGFE n = 3, and TcdBFZD−/CSPG4− n = 3, TcdBCSPG4− n = 3) (c), and pathological scores (error bars indicate mean ± SEM, PBS n = 4, TcdB n = 5, TcdBGFE n = 5, and TcdBFZD−/CSPG4− n = 5, TcdBCSPG4− n = 5) (d). p values were calculated by post hoc analysis of a by one-way ANOVA using Holm-Sidak’s test for multiple comparisons: ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05. The exact p values are presented in the accompanying source data.
We analyzed the toxicity of these TcdB1 mutants in comparison with the WT toxin by directly inject them into the mouse cecum5b–d and Supplementary Fig. 8g). Both TcdBGFE and TcdBCSPG4− showed greatly reduced potency, with no significant difference between them: both showed modest levels of inflammatory cell infiltration and submucosal edema, and mild to normal levels of disruption of epithelium, tight junction, and hemorrhagic congestion. TcdBFZD−/CSPG4− showed further reduced toxicity, with minimal levels of disruption to cecum tissues under our assay conditions (Fig. 5d and Supplementary Fig. 8g). These results demonstrate that FZDs and CSPG4 act as independent receptors in TcdB1 pathogenesis in vivo.
Bezlotoxumab is the only FDA-approved therapeutic antibody against TcdB, and a prior study suggested that bezlotoxumab reduced binding of TcdB to CSPG4 in vitro in immunoprecipitation assays9a)6a)6b). Since CSPG4 binds TcdB by simultaneously interacting with the CPD, DRBD, hinge, and CROPs, bezlotoxumab binding may reorient the CROPs relative to the rest of TcdB and compress the CSPG4-binding groove, thus preventing CSPG4 binding in an allosteric manner (Fig. 6b).
a A structure model showing the binding of CSPG4 and bezlotoxumab (PDB: 4NP4) in TcdB holotoxin (PDB: 6OQ5). TcdB holotoxin and CSPG4 Repeat1 are showing as surface models with the GTD, CPD, DRBD, CROPs, and CSPG4 Repeat1 colored in pink, blue, orange, cyan, and green, respectively. The two Fab fragments of bezlotoxumab are shown as cartoon models and colored blue and purple. E1 and E2 indicate the epitope-1 and epitope-2 for bezlotoxumab in TcdB. A close-up view into the conflicting area between the Fab 1 bound at the E1 site and TcdB is shown in a red oval box, while the Fab residues that sterically clash with TcdB are colored in green. b A proposed model for allosteric interactions between CSPG4 and bezlotoxumab (Bezlo). c TcdB1 could not bind CSPG4mini when it was prebound to the immobilized bezlotoxumab according to BLI assays. d Bezlotoxumab could still bind TcdB1 when it was prebound to the immobilized CSPG4 Repeat1. Sequential loading of different proteins to the biosensor is indicated by different background colors. e The protection effects of inhibitors against TcdB1 and TcdB2 were quantified by the cytopathic cell-rounding assay on HeLa cells. HeLa cells were incubated with TcdB1 (10 pM) or TcdB2 (100 pM) in the presence of serial-diluted bezlotoxumab (bezlo), its Fab (Fab), or Repeat1-Fc (Repeat1). Percentage of rounded cells are plotted by inhibitor concentrations at 6 h. Error bars indicate mean ± sd (n = 3 biologically independent experiments). f, g The protective effects of Repeat1-Fc and bezlotoxumab against TcdB1 and TcdB2 were examined in vivo using the cecum injection assay. TcdB1 (6 µg), TcdB2 (6 µg), TcdB1 or TcdB2 with Repeat1-Fc (30 µg) or bezlotoxumab (52 µg), Repeat1-Fc alone (30 µg), or the PBS control was injected into the cecum of CD1 mice in vivo. The cecum tissues were harvested 6 h later, and the representative H&E staining (scale bar represents 100 µm, PBS n = 5, B1 n = 13, B1 + Repeat1 n = 6, B1 + Bezlo n = 6, B2 n = 15, B2 + Repeat1 n = 7, B2 + Bezlo n = 6, Repeat1 n = 4) (f) and the histological scores (error bars indicate mean ± SEM, PBS n = 5, B1 n = 13, B1 + Repeat1 n = 6, B1 + Bezlo n = 6, B2 n = 15, B2 + Repeat1 n = 7, B2 + Bezlo n = 6, Repeat1 n = 4) (g) are shown. p values were calculated by post hoc analysis of a one-way ANOVA using Holm-Sidak’s multiple comparison test: ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05. The exact p values are presented in the source data.
To verify this hypothesis, we examined the competition between bezlotoxumab and CSPG4 using BLI and pull-down assays. We found that when TcdB1 and TcdB2 were prebound with the immobilized bezlotoxumab, CSPG4 could not bind subsequently (Fig. 6c and Supplementary Fig. 9b, d). Meanwhile, the CSPG4-bound TcdB1 and TcdB2 could still bind bezlotoxumab, which is likely due to single-site antibody binding to epitope-2 (Fig. 6d and Supplementary Fig. 9c, e). To further understand how the single- vs. double-epitope binding modes effect bezlotoxumab’s activity, we examined the neutralization potency against TcdB1 for bezlotoxumab and its Fab fragment using the cell-rounding assay. When antibodies were preincubated with TcdB1 (10 pM) before adding to the culture medium, bezlotoxumab completely protected cells within 6 h at the lowest concentration tested (16 nM), but its Fab did not show any protection until the concentration reached 2 µM, which only reduced cell rounding by ~40% (Fig. 6e and Supplementary Fig. 10a, b). We believe this is due to the lack of synergy on TcdB binding between individual Fab molecules. These data consistently define a unique mechanism for bezlotoxumab at the molecular level, where it relies on synergistic binding to both epitopes in TcdB using its two Fab arms.
We further evaluated Repeat1-Fc and bezlotoxumab for blocking TcdB1 and TcdB2 in vivo using the mouse cecum injection model. Briefly, TcdB1 or TcdB2 (6 µg) was preincubated with Repeat1-Fc (30 µg) or bezlotoxumab (52 μg), respectively, and the mixture was injected into the mouse cecum. The cecum tissues were dissected out for histological analysis 6 h later. As shown in Fig. 6f, g and Supplementary Fig. 10f, Repeat1-Fc was able to reduce overall damage to cecum tissues from both TcdB1- and TcdB2-treated mice, including less inflammatory cell infiltration, submucosal edema, hemorrhagic congestion, and epithelial disruption, while bezlotoxumab was only effective in reducing TcdB1 toxicity, but showed no effect on TcdB2 under the same assay conditions.
Biomedical scientists are dreaming of a technique showing the distribution of all the biomolecular constituents that make up biological tissue in high-resolution, three-dimensional maps. Such a visualization tool does not currently exist. Whereas fluorescence and refraction based techniques will never be able to identify an arbitrary molecular compound in tissue, vibrational imaging techniques offer much more promise. They work label-free and non-invasively and they are able to identify many important groups.
The conventional vibrational imaging technique is Raman microscopy. Practical limitations have so far prevented Raman microscopy from reaching its full potential. The most important limitation is speed. The intrinsically weak Raman signals severely limit the achievable image acquisition rate. The development of coherent Raman scattering (CRS) microscopy techniques over the last decade has resulted in important steps toward resolving the speed issue. In a feature article, Eric O. Potma and a team of scientists at the University of California (Irvine, USA) discuss several ways in which the improved speed of CRS microscopy has transformed the biomedical imaging field and touch on some of the challenges that lie ahead in moving towards the realization of a more generally applicable visualization technique.
Stronger signals are obtained because in CRS the molecules are driven coherently, which makes them radiate in unison. The resulting signal is coherently amplified through constructive interference in a well-defined, phase-matched direction which enables efficient detection of the Raman response. For instance, when the microscopic focal volume is filled with lipids, the number of detected photons in coherent anti-Stokes Raman scattering (CARS), generated from the CH2 stretching mode, can easily exceed 102 per microsecond at 10 mW of illumination. With such high signal levels, real-time Raman imaging of biological tissues becomes feasible.
The intrinsic movement of the tissue, due to pulsation of blood vessels, breathing, or positional adjustments by the subject, poses a challenge when imaging living tissues. Imaging at video-rate enables image acquisition in which the individual frames show minimal blurring due to movements. Another advantage of fast imaging is the reduction of possible photodamage in living tissues. Besides live tissue imaging in small animal models, CRS imaging has already been applied to the examination of human skin in vivo.



 8613371530291
8613371530291